Wiedza teoretyczna
INFORMACJE PODSTAWOWE
Zespół Angelmana (ang. Angelman Syndrome, AS) jest chorobą o podłożu genetycznym związaną z zaburzeniem rozwoju i funkcji ośrodkowego układu nerwowego.
CZĘSTOŚĆ WYSTĘPOWANIA
Około 1/15000 urodzeń na całym świecie (około 20 wszystkich urodzeń w Polsce).
DŁUGOŚĆ ŻYCIA
Średnia długość życia chorych jest normalna, jednak do końca życia nie są oni samodzielni.
OBJAWY
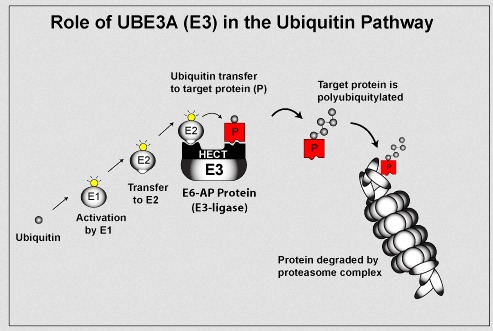
Objawy zespołu Angelmana wynikają z zakłócenia funkcji genu UBE3A, kodującego enzym – ligazę ubikwityny. Gen ten jest zlokalizowany w regionie 15q11.2-q13. Ligaza ubikwityny E3A jest enzymem służącym do oznaczenia uszkodzonych białek w komórkach, które powinny zostać zniszczone. Struktury komórkowe określane jako proteasomy, rozpoznają i degradują białka oznaczone ubikwityną. Proces degradacji białek w komórce jest bardzo ważny, ponieważ usuwa on zbędne i uszkodzone białka, co zapewnia prawidłowe funkcjonowanie komórek. Badania sugerują, że ligaza ubikwityny odgrywa dużą rolę w prawidłowym rozwoju i funkcjonowaniu układu nerwowego.
Źródło:
https://www.peds.ufl.edu/divisions/genetics/programs/angelman_syndrome/ube3a_gene.htm
PRZYCZYNY
Ludzie zwykle dziedziczą dwie kopie genu UBE3A, po jednym od każdego z rodziców. Obie kopie tego genu są włączone, czyli aktywne, w większości tkanek organizmu. W niektórych obszarach mózgu, tylko kopia genu odziedziczona od matki (kopia matczyna) jest aktywna, natomiast kopia genu odziedziczona od ojca jest nieaktywna. Jest to stan prawidłowy.
Jednakże w przypadku zespołu Angelmana, w mózgu zachodzi tzw. preferencyjna ekspresja allelu (tj. kopii genu) matczynego i stąd mówi się, że defekt ten jest pochodzenia matczynego.
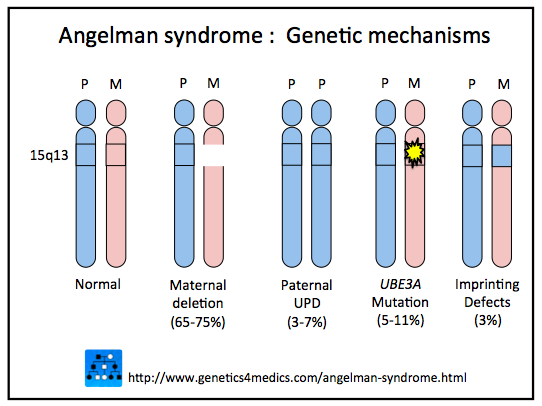
Wyróżnia się 4 główne mechanizmy, które zakłócają funkcję tego genu, a przez to uważane są za podłoże zespołu Angelmana:
1. Delecja (wypadnięcie) (ang. Maternal Deletion) fragmentu chromosomu 15 w regionie 15q11-q13 pochodzenia matczynego. Typowe delecje są duże, zawierają około 6 milionów par zasad. Delecja występuje w około 65-75 % przypadków. Ta sama delecja jest przyczyną zespołu Pradera – Williego, z tą różnicą, że wtedy defekt pochodzi z chromosomu ojcowskiego.
2. Disomia uniparentalna (UPD) (ang. Paternal UPD) – czyli nieprawidłowy stan, w którym obie kopie genów danego chromosomu są odziedziczone po jednym z rodziców (w przypadku AS – po ojcu). Wykazano, że ta forma mutacji wiąże się z lżejszym przebiegiem zespołu Angelmana – np. rzadszymi napadami padaczkowymi. Disomia występuje w około 3-7% przypadków.
3. Mutacja genu UBE3A (ang. UBE3A Mutation) – dotyczy około 5-11% przypadków.
4. Defekt imprintingu (ang Imprinting Defects) – czyli zaburzenie w procesie dołączania reszt metylowych który ma miejsce podczas wytwarzania gamet. Dotyczy około 3% przypadków.
http://genetics4medics.com/angelman-syndrome.html
KORELACJA GENOTYPOWO-FENOTYPOWA
Wszystkie z wymienionych mechanizmów genetycznych mogą prowadzić do wystąpienia zbliżonego obrazu klinicznego z ciężką i głęboką niepełnosprawnością intelektualną, charakterystycznym zachowaniem i ciężkimi zaburzeniami mowy. Można dostrzec pewne cechy wspólne dla poszczególnych genotypów. Te korelacje są przedstawiono poniżej:
- Delecje są najsilniej wyrażone w postaci małogłowia, napadów padaczkowych, względnie też w zmianach w pigmentacji skóry, włosów i tęczówek, zaburzeniach ruchowych (np. ataksji, zmniejszeniu napięcia mięśniowego, problemami w karmieniu) oraz obniżonymi zdolnościami poznawczymi i językowymi.
- Dzieci z UPD i ID mogą wykazywać mniej cech dymorficznych (np. rzadziej małogłowie), mniej zaburzeń ruchowych, w tym ataksji a także mają mniejszą częstość (ale nie brak) napadów padaczkowych.
- Dzieci z ID wykazują lepsze zdolności poznawcze, językowe i motoryczne. Najbardziej zaawansowane zdolności mowy występują w grupie ID, który jest mozaiką z ID bez delecji (około 20% grupy ID). Osoby te mogą opanować do 50-60 słów i używać prostych zdań.
- Grupa mutacji UBE3A jest pośrednia między delecją a ID pod względem małogłowia, drgawek, trudności motorycznych oraz umiejętności językowych. Niektóre dzieci z UBE3A mogą mieć stosunkowo wysokie zdolności poznawcze, dobrą sprawność ruchową oraz umiejętności motoryczne. Jest to przypuszczalnie efekt jakości ich mutacji (np. lokalizacja i rodzaj zmian DNA w obrębie genu) co powoduje mniej poważnych problemów klinicznych.
BADANIA GENETYCZNE
Diagnostyka molekularna zespołu obejmuje :
- analizę chromosomową w poszukiwaniu dużych delecji oraz mikrodelecji (FISH)
- analizę metylacji w badanym regionie DNA
- analizę polimorfizmu markerów mikrosatelitarnych
- poszukiwanie mutacji w genie UBE3A
Celem badań jest weryfikacja rozpoznania klinicznego i ustalenie rodzaju defektu molekularnego. Badanie FISH jednoznacznie wskazuje na brak lub obecność delecji. Nowe metody diagnostyczne, takie jak technika porównawczej hybrydyzacji genomowej, potrafią ukazać rozległość delecji, podczas gdy FISH tego nie potrafi. Duże delecje powodują całkowite usunięcie genu UBE3A z chromosomu matki, ich zasięg może także powodować braki w dalszych częściach chromosomu, jak na przykład w genach kodujących receptor GABA. Ale to głównie brak UBE3A daje objawy kliniczne i problemy związane z AS.
Badanie w kierunku AS można wykonać w większości Poradni Genetycznych w Polsce.
CECHY CHARAKTERYSTYCZNE
Do cech charakterystycznych tego zespołu należą:
- opóźnienie w rozwoju psychoruchowym i intelektualnym (trudności w nauce, rozumieniu poleceń i powtarzaniu),
- zaburzenia rozwoju mowy – dzieci te często w ogóle nie mówią, co najwyżej używają kilku/kilkunastu słów,
- zaburzenia ruchu i równowagi, trudności w poruszaniu się, z ataksją chodu i drżeniem kończyn, tłumacząc to najprościej dzieci z zespołem Angelmana nie czują, że utrzymują równowagę,
- częste uśmiechanie się i napady śmiechu, łatwa ekscytacja,
- nadpobudliwość, problemy z koncentracją i skupianiem uwagi.
Do równie częstych objawów tej choroby należą:
- napady padaczkowe (zwykle pojawiają się przed 3 rokiem życia),
- wysunięty język, wypychanie języka z ust,
- zaburzenia ssania i połykania (trudności w karmieniu w okresie niemowlęcym),
- częste ślinienie się, wkładanie przedmiotów do ust,
- przygięte ramiona (zwłaszcza podczas chodzenia),
- machanie rączkami,
- zainteresowanie sznurkami, kabelkami,
- zaburzenia snu,
- zainteresowanie i fascynacja wodą.
OPIS KLINICZNY I PRZEBIEG CHOROBY
1. Noworodki
Dzieci dotknięte zespołem Angelmana najczęściej rodzą się z ciąży i porodu o niepowikłanym przebiegu, z prawidłową długością i masą ciała ( z reguły jednak niższą o około 200-300g od masy urodzeniowej zdrowych dzieci) oraz pozostającymi w granicach normy obwodami główki i klatki piersiowej.
U ok. 70% chorych występują zaburzenia odżywiania. Obserwuje się trudności w ssaniu (dotyczy to zwłaszcza karmienia naturalnego), wymioty po posiłkach (często przez nos), oraz mały przyrost masy ciała. Krztuszenie się pokarmem (niekiedy rozpoznaje się refluks żołądkowo-przełykowy) często prowadzi do zaburzeń oddechowych. U ok. 15% chorych noworodków wymienione zaburzenia są przyczyną hospitalizacji.
W tym wczesnym okresie życia dzieci te sprawiają niekiedy wrażenie nadmiernie spokojnych. Są wiotkie, przy trzymaniu na rękach wyczuwa się drżenie ich ciała, u niektórych z nich występuje, wkrótce zresztą ustępujący, oczopląs.
2. Dzieci
Ewidentne opóźnienie rozwoju psychoruchowego obserwuje się dopiero ok. 6 m.ż., u niektórych chorych rozwój psychoruchowy do końca 1 r.ż. wydaje się być prawidłowy. Inne objawy ujawniają się stopniowo we wczesnym dzieciństwie. Zespół Angelmana jest rzadko rozpoznawany u bardzo małych dzieci. Najczęściej pierwszą diagnozę stawia się miedzy 2 a 5 rokiem życia, kiedy charakterystyczne objawy fenotypowe i zachowanie stają się silniej wyrażone.
U 100% dzieci:
- opóźnienie w rozwoju psychoruchowym
- zmiany w zachowaniu
- opóźnienie w nabywaniu umiejętności mowy
Opóźnienie rozwoju ruchowego spowodowane jest: wiotkością mięśni tułowia, nadmiernym napięciem mięśniowym w kończynach dolnych oraz zaburzeniami równowagi. Przejawiają się zwykle jako ataksja i/lub drżący ruch kończyn. Zaburzenia ruchowe mogą być łagodne, mogą przejawiać się jako drżenie, niezborność ruchów lub szybkie, mimowolne ruchy.
Dzieci z AS siadają samodzielnie pomiędzy 1 a 3 r.ż. Siedzą dość charakterystycznie, podpierając się jedną ręką z powodu osłabienia mięśni tułowia: kończyny dolne, w których występuje nadmierne napięcie mięśniowe, mają natomiast szeroko rozsunięte i sztywne. Około 22 m.ż. rozpoczyna się nietypowe raczkowanie, które przypomina czołganie się bądź przemieszczają się posuwając na pośladkach. Na ogół nie lubią pozycji leżącej na brzuchu, jednak niektóre z nich z tej właśnie pozycji, opierając sztywne kończyny dolne o podłoże, usiłują dźwigać się ku górze na ramionach. Chodzić samodzielnie zaczynają między 18 m. a 7 r.ż. ,przeważnie od 5 r.ż. Chód jest bardzo charakterystyczny: chwiejny (ataktyczny), na szerokiej podstawie i sztywnych kończynach dolnych. Około 30% chorych nie chodzi. Najczęstszą przyczyną takiego stanu jest skolioza czyli boczne skrzywienie kręgosłupa (często obecne od urodzenia, oporne na leczenie), znaczna wiotkość tułowia i nasilona spastyczność kończyn dolnych. Większość nierehabilitowanych systematycznie chorych ok. 20 r.ż. nie chodzi z powodu utrwalenia się przykurczów w stawach kolanowych oraz znacznego stopnia skrzywienia kręgosłupa.
Jednym z najbardziej charakterystycznych objawów jest fenotyp behawioralny (zachowanie). Osoby z AS sprawiają wrażenie szczęśliwych, są towarzyskie, często się uśmiechają.
Pierwszy uśmiech pojawia się we właściwym czasie (4 – 6 tydz. ż.), występuje często niekiedy (ok. 10 tydz. ż.) w postaci łatwego do sprowokowania chichotu. Napady śmiechu nie są warunkiem niezbędnym do rozpoznania tego zespołu, jednak ich obecność (u ok. 96% chorych) bardzo pomaga w jego ustaleniu. Śmiech jest nieadekwatny do sytuacji (np. występuje w czasie zastrzyków), bardzo łatwy do wywołania i nie poddaje się kontroli. W młodszym wieku napadom śmiechu towarzyszy machanie bądź klaskanie rękami.
Dzieci te fascynują się m.in. wodą, pofałdowanymi przedmiotami, takimi jak niektóre papiery i tworzywa sztuczne. Bardzo lubią lejącą się z kranu wodę, kąpiele, załamujące się refleksy świetlne oraz hałas (chętnie bawią się zabawkami wydającymi dźwięki).
Większość chorych dzieci ma również problemy ze snem i potrzebują mniej snu niż ich zdrowi rówieśnicy, co stanowi poważny problem dla zdrowia rodziny.
Objawem charakterystycznym są też zaburzenia mowy. Objawiają się jako brak lub minimalne wykorzystanie słów. Natomiast duża jest zdolność do niewerbalnych umiejętności komunikacyjnych. Około 30% dzieci nie mówi w ogóle, pozostałe wymawiają zaledwie pojedyncze słowa, najczęściej: mama, tata, baba. Porozumiewają się z rodzicami za pomocą wskazywania, niekiedy określonych gestów lub pociągania za rękę bądź odzież. Z tego powodu, możliwie jak najwcześniej należy umożliwić dzieciom korzystanie ze specjalnych pomocy pozwalających rozwijać komunikację alternatywną.
Pozostałe cechy:
U ponad 80% dzieci
- opóźniony, nieproporcjonalny przyrost obwodu głowy, co skutkuje zwykle stwierdzeniem małogłowia (≤ 2 SD normalnej OFC) w wieku 2 lat. Małogłowie jest większa u osób z delecją 15q11.2-q13. Krótkogłowie i małogłowie (poniżej 25 centyla) u dzieci powyżej 2 r.ż. należą do objawów stałych.
- Napady padaczkowe z nieprawidłowościami w EEG
Cechy skojarzone (20 – 80%)
- Twarz – Cechy dysmorficzne twarzy mogą mieszać się z cechami urody danej rodziny, co może utrudniać ich wychwycenie. Twarz, którą określa się obecnie jako „twarz Angelmana” w okresie noworodkowym i niemowlęcym nie wykazuje żadnej odrębności, natomiast wyraźnie zmienia się wraz z wiekiem dziecka i z czasem wykazuje coraz więcej cech charakterystycznych. Zmiany spowodowane są przede wszystkim stałym wysuwaniem języka (dziecko prawie zawsze trzyma język pomiędzy zębami, bądź, w późniejszym wieku, wykłada język na wargi, a nawet na brodę), oraz postępującym prognatyzmem. Przypuszcza się, że do wystąpienia wyżej opisanych zaburzeń predysponuje mała żuchwa i strome ustawienie podstawy czaszki.Środkowa część twarzy jest hipoplastyczna, rysy twarzy delikatne. Oczy osadzone są głęboko, stosunkowo szeroko, niekiedy zaznaczone są zmarszczki nakątne. Zęby są rozstawione szeroko, a u starszych dzieci, na skutek ucisku wywieranego przez stale wysuwany język, powierzchnie górne koron zębów są łukowato wycięte (starte). Usta są bardzo duże, warga górna wąska. Bródka jest wydatna i ok. 5 r.ż. dolna część twarzy staje się szeroka. Mimo tych odrębności, dzieci są bardzo podobne do swoich rodziców.
- Szkielet i kości
- Płaska potylica
- U ok. 35% stwierdza się poziome zagłębienie (bruzdę) w okolicy potylicznej.
- Skolioza (boczne skrzywienie kręgosłupa)
- Pigmentacja skóry i włosów – U ok. 70% chorych obserwuje się hipopigmentację skóry, włosów i oczu (dotyczy to dzieci z udokumentowaną za pomocą badań molekularnych delecją regionu 15q11-q13). Włosy są jasne, skóra jasna i wrażliwa na promieniowanie słoneczne. W warstwie przedniej zrębu tęczówki oraz w naczyniówce stwierdza się niedobór barwnika.
- Zaburzenia okulistyczne – U 42% występuje zez i umiarkowanego stopnia krótkowzroczność.
3. Dorośli
Główne problemy dorosłych osób z AS są w istocie kontynuacją problemów występujących w dzieciństwie. Młodzi dorośli z zespołem Angelmana robią postępy kiedy są poddawani stałej terapii i nauce. Nie wykazują tendencji do cofania się w postępach. Ich zdrowie fizyczne jest dość dobre.
Do głównych problemów zdrowotnych charakterystycznych dla dorosłych z AS należą problemy kontroli napadów padaczkowych, zaburzenia zachowania i zaburzenia ruchowe. Napady padaczkowe z wiekiem mają mniejsze nasilenie i częstość. Nie stwierdza się dużej neurodegeneracji ani widocznych zmian morfologicznych w badaniu MRI głowy (mózgowia), mimo chronicznych napadów. Większym problemem natomiast staje się poruszanie. Problemy mogą wynikać ze skoliozy i ataksji. Skolioza najczęściej rozwija się w wieku nastoletnim, powinna być wcześnie wychwycona i wcześnie leczona żeby zapobiec progresji. W cięższych przypadkach może wymagać leczenia operacyjnego.
Dorośli z AS, z powodu problemów behawioralnych, mogą mieć podawany lek neuroleptyczny a skutki uboczne lub uspokajające działanie tych leków może stanowić kolejny problemem zdrowotny.
Wiele problemów zdrowotnych jest właściwie takich samych jak te spotykane u osób niedotkniętych AS. Wraz z wiekiem osoby z zespołem Angelmana stają się mniej pobudliwe i maleją problemy ze snem.
Nie stwierdzono zwiększonej predyspozycji do wystąpienia nowotworów stąd długość życia może być tylko nieznacznie niższa od średniej. Istnieją doniesienia o osobach żyjących z AS ponad 70 lat, choć nie ma na razie żadnych udokumentowanych danych które szacują żywotność.
Dorośli dotknięci zespołem Angelmana są zależni od opiekunów przez całe życie, aczkolwiek próbuje się stworzyć mieszkania i miejsca pracy dostosowane do potrzeb osób z AS.
DIAGNOSTYKA RÓŻNICOWA
W diagnostyce różnicowej, zwłaszcza w okresie niemowlęcym, należy uwzględnić :
- zespół Retta
- zespół Pitta-Hopkinsa
- zespół Lennoxa-Gastauta
- autyzm niemowlęcy
- zespół Mowat-Wilsona
PORADNICTWO GENETYCZNE
Większość przypadków zespołu Angelmana nie jest dziedziczna a w szczególności te, które wynikają z delecji genu UBE3A w matczynym chromosomie 15 lub disomią (UPD) chromosomu 15. Te zmiany maja charakter przypadkowy.
Ryzyko powtórzenia choroby u kolejnego dziecka gdy u pierwszego stwierdzono AS:
- Delecję de novo wynosi <1%
- UPD wynosi <1%
Rzadko zmiany genetyczne odpowiedzialne za zespół Angelmana mogą być dziedziczone.
Na przykład możliwe jest dziedziczenie mutacji w genie UBE3A lub dziedziczenie zmian w DNA dezaktywujących matczyny gen UBE3A.
Matki nosicielki zrównoważonej aberracji obejmującej region 15q11-q13 mają podwyższone ryzyko urodzenia kolejnego chorego dziecka.
DIAGNOSTYKA PRENATALNA
Ewentualna diagnostyka prenatalna może zostać przeprowadzona w szczególnych przypadkach z wykorzystaniem techniki FISH bądź analizy metylacji.
ZALECANE KONSULTACJE
1. Noworodki i dzieci
KONSULTACJA NEUROLOGICZNA
Głównie w zakresie kontroli i leczenia napadów padaczkowych.
KONSULTACJA GASTROENTEROLOGICZNA
W przypadku znacznych problemów z karmieniem dziecko powinno być pod opieką gastroenterologa dziecięcego. Celem konsultacji jest wykluczenia refluksu żołądkowo-przełykowego oraz oceny, czy dziecko ssie i połyka prawidłowo.
Przy problemach z ulewaniem, wymiotami zastosowanie zagęszczonych mieszanek mleka modyfikowanego (oznaczonych symbolem „AR”) oraz prawidłowe układanie dziecka po posiłku może złagodzić objawy refluksu.
KONSULTACJA OKULISTYCZNA
Częste wady i choroby okulistyczne wymagają dokładnej oceny specjalistycznej pacjentów z AS i ewentualnej dalszej opieki w przypadku stwierdzenia nieprawidłowości. U prawie połowy pacjentów z AS występuje zez i umiarkowanego stopnia krótkowzroczność.
KONSULTACJA ORTOPEDYCZNA
Z powodu często współistniejącej skoliozy, czasem wymagającej korekty chirurgicznej.
Ważne jest wczesne rozpoczęcie leczenia aby zapobiec progresji choroby.
KONSULTACJA LOGOPEDYCZNA
Z powodu problemów z mową i słabym jej rozwojem.
Kierunki pracy logopedycznej:
- usprawnianie narządów mowy,
- wzbogacenie zasobu słownictwa,
- rozpatrzenie możliwości równoczesnego wprowadzenia alternatywnej formy komunikacji np. obrazkowej.
ALTERNATYWNE I WSPOMAGAJĄCE METODY KOMUNIKACJI
(Metody komunikowania omówiono na podstawie książki J. Błeszyński (red) „Alternatywne i wspomagające metody komunikacji.” Oficyna Wydawnicza „Impuls”, Kraków 2006).
Osoby z zespołem Angelmana nie są w stanie porozumiewać się przy wykorzystaniu klasycznego sposobu komunikacji w postaci mowy dźwiękowej. Dlatego potrzebują wyjątkowej uwagi, wyjątkowego wsparcia oraz odmiennych, alternatywnych, zastępczych, dostosowanych do indywidualnych możliwości i predyspozycji metod komunikacji. Do naszej rodzimej pedagogiki specjalnej, dzięki zaangażowaniu i wysiłkowi wielu specjalistów, wprowadzane są nowe rozwiązania w zakresie pomocy nawiązywania, utrzymywania i doskonalenia interakcji komunikacyjnych w sposób wspomagający i alternatywny, tj. wykorzystujący gest, obraz, symbol graficzny. Metody te stwarzają dziecku warunki do ekspresji potrzeb, dokonywania wyborów, zadawania pytań, przeczenia, prowadzenia rozmowy.
- Piktogramy
Systemem znaków obrazkowych służących do rozwijania komunikacji dla osób z niepełnosprawnością umysłową i fizyczną oraz dla osób z poważnymi problemami w zakresie rozumienia języka i posługiwania się mową dźwiękową.
- System Komunikacji Symbolicznej Bliss
Metoda porozumiewania się, w której słowa przedstawione są w postaci rysunku.
- Gesty oraz systemy gestów
KONSULTACJA PSYCHOLOGICZNA I PEDAGOGICZNA
Ma na celu:
- rozwijanie umiejętności koncentrowania i utrzymywania uwagi na istotnych bodźcach,
- wdrażanie do respektowania prostych zakazów i nakazów,
- wdrażanie do celowych działań zabawowych i samoobsługowych (na miarę możliwości ruchowych),
- objęcie zajęciami metody integracji sensorycznej
Terapia pedagogiczna
- Metoda Ruchu Rozwijającego Weroniki Sherborne
- Malowanie dziesięcioma palcami
- Dogoterapia
- Hipoterapia
- Program Aktywności Knill
- Metoda Felicie Affolter
ZABIEGI REHABILITACYJNE I TERAPEUTYCZNE
Mają na celu:
- stymulację reakcji równoważnych;
- poprawę koordynacji ruchowej;
- naukę prawidłowych wzorców ruchowych;
- normalizację wzorców postawy.
Rehabilitacja psychoruchowa:
- Metoda Bobath’ów
Metoda neurorozwojowego leczenia usprawniającego Karela i Berty Bobath polega na hamowaniu patologicznych odruchów i nieprawidłowych wzorców ruchowych oraz rozwijaniu czynności ruchowych w kolejności, w jakiej pojawiają się one w rozwoju dziecka zdrowego .
- Ćwiczenia w wodzie
Ćwiczenia w wodzie są jedną z najkorzystniejszych form zajęć z dziećmi, szczególnie z dziećmi z zespołem Angelmana, u których występuje fascynacja wodą. Już samo wejście do wody sprawia większości wiele radości i zadowolenia.
2. Dorośli
Konsultacje u dorosłych:
Jak wyżej. Główne problemy dorosłych u osób z AS są w istocie kontynuacją problemów występujących w dzieciństwie.
ZASOBY INTERNETOWE
http://www.angelman.org
http://ghr.nlm.nih.gov/condition/angelman-syndrome/
http://omim.org/entry/105830
http://www.orpha.net
http://www.angelmantoday.com/understanding-genetic-classes/
https://angelmannetwork.wordpress.com/angelman-syndrome/about-angelman-syndrome/
http://bioinfo.mol.uj.edu.pl/articles/Sikora05
http://www.eduteka.pl/doc/zespol-angelinami
http://www.osw.wyszkow.com.pl/wp-content/gazetka-nr-6..pdf
http://bioinfo.mol.uj.edu.pl/articles/Zieba04″
Dodaj komentarz
Musisz się zalogować, aby móc dodać komentarz.